
Picky v. 3.0

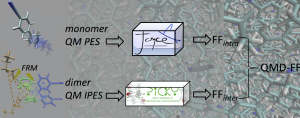
The interaction energies obtained by Quantum Mechanical (QM) calculations for a number of selected dimer conformations extracted from classical Molecular Dynamics (MD) simulations are used to parameterize inter-molecular force fields, through an iterative automated procedure.
The Picky package is a collection of computational tools devised to obtain Quantum Mechanically Derived Force-Fields (QMD-FF) through an automated protocol, which integrates accurate quantum mechanical calculations with classical numerical simulations. Intermolecular FF parameters are obtained by an automated iterative approach, aimed to minimize the difference between the interaction potential energy surfaces (IPESs) obtained by QM calculations and employing the QMD-FF. The IPESs are sampled through a large number of dimers in different arrangements, which are automatically extracted by Picky from equilibrated conformations generated by MC or MD runs.
The package consists in several programs: the main Picky code, which can be used alone to perform dimer sampling over a given configuration, the Pickyfit code, to obtain FF parameters through a least-square non linear fitting procedure against QM data, the Pickyrecover code, which retrieves intermolecular energies and geometries from the QM database and a number of other utilities to assist the user during the parameterization.
Intermolecular FFs, obtained with thePicky procedure, can be further coupled with an intramolecular part to achieve a more realistic description of the system. In such case, a fully QM derived FF can be set up by adopting the Joyce protocol for the intra-molecular part.
All codes were written in Fortran by Giacomo Prampolini, Ivo Cacelli, Antonella Cimoli, and Paolo Roberto Livotto.
All Picky software is free and can be redistributed and/or modified under the terms of the GNU General Public License as published by the Free Software Foundation (version3).
For further information please contact Giacomo Prampolini.
Lastest distributed version
Fill the following form to receive the download link.
Installation and user guide
Test suite with example files
Previous versions (available upon request):
Picky version 2.3 (route I and route II implementation, picky assistant automation, 2015, [2])
Picky version 2.5 (virtual sites implementation, placevirt utility, 2019, [3)]
Selected application performed by the authors.
Liquid Crystals – predicting material properties through accurate QMD-FFs
LC topologies & related Gromacs files
da Silveira, L.G., Livotto, P.R., Padula D., Vilhena, J.G., Prampolini, G. “Accurate Quantum-Mechanically Derived Force-Fields through a Fragment-Based Approach: Balancing Specificity and Transferability in the Prediction of Self-Assembly in Soft Matter”, J. Chem. Theor. and Comput., 18, 6905 (2022).
https://pubs.acs.org/doi/10.1021/acs.jctc.2c00747
Prampolini, G., da Silveira, L.G., Vilhena, J.G., Livotto, P.R “Predicting Spontaneous Orientational Self-Assembly: In Silico Design of Materials with Quantum Mechanically Derived Force Fields”, J. Phys. Chem. Lett., 13, 243 (2021).
https://pubs.acs.org/doi/10.1021/acs.jpclett.1c03517
Vilhena, J.G., da Silveira, L.G., Livotto, P.R., Cacelli, I., Prampolini, G. “Automated Parameterization of Quantum Mechanically Derived Force Fields for Soft Materials and Complex Fluids: Development and Validation”, J. Chem. Theor. and Comput., 17, 4449 (2021).
References
Method’s development:
[1] I. Cacelli, A. Cimoli., P.R. Livotto, and G. Prampolini, G, “An automated approach for the parameterization of accurate intermolecular force-fields: Pyridine as a case study” J. Comp. Chem. , 33, 1055–1067 (2012)
[2] G. Prampolini, P.R. Livotto, and I. Cacelli, “Accuracy of Quantum Mechanically Derived Force-Fields Parameterized from Dispersion-Corrected DFT Data: The Benzene Dimer as a Prototype for Aromatic Interactions” J. Chem. Theory Comput., 11, 5182–5196 (2015) [3] M. Campetella,N. De Mitri, and G. Prampolini “Automated parameterization of quantum-mechanically derived force-fields including explicit sigma holes: A pathway to energetic and structural features of halogen bonds in gas and condensed phase.” J. Chem. Phys., 153, 044106 (2020)
Selected applications:
[4] G. Prampolini, M. Campetella, N. De Mitri, P.R. Livotto and I. Cacelli “Systematic and Automated Development of Quantum Mechanically Derived Force Fields: The Challenging Case of Halogenated Hydrocarbons.” J. Chem. Theor. Comput., 12, 5525–5540 (2016)
[5] L. Greff da Silveira, M. Jacobs, G. Prampolini, P.R: Livotto, and I. Cacelli “Development and Validation of Quantum Mechanically Derived Force-Fields: Thermodynamic, Structural, and Vibrational Properties of Aromatic Heterocycles” J. Chem. Theor. Comput., 14, 4884–4900 (2018)