
Picky v. 3.0

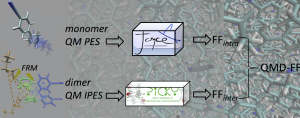
Le energie di interazione ottenute dai calcoli di Meccanica Quantistica (QM) per un numero di conformazioni dimeriche selezionate estratte dalle classiche simulazioni di Dinamica Molecolare (MD) vengono utilizzate per parametrizzare i campi di forza intermolecolari, attraverso una procedura automatizzata iterativa.
Il pacchetto Picky è una raccolta di strumenti computazionali ideati per ottenere campi di forza derivati quanto-meccanicamente (QMD-FF) attraverso un protocollo automatizzato, che integra calcoli di meccanica quantistica accurati con simulazioni numeriche classiche. I parametri FF intermolecolari sono ottenuti mediante un approccio iterativo automatizzato, volto a minimizzare la differenza tra le superfici di energia potenziale di interazione (IPES) ottenute dai calcoli QM e impiegando il QMD-FF. Gli IPES vengono campionati attraverso un gran numero di dimeri in diverse disposizioni, che vengono estratti automaticamente da Picky da conformazioni equilibrate generate da corse MC o MD.
Il pacchetto è composto da diversi programmi:
il codice Picky principale, che può essere utilizzato da solo per eseguire il campionamento del dimero su una determinata configurazione;
il codice Pickyfit, per ottenere i parametri FF attraverso una procedura di fitting non lineare dei minimi quadrati rispetto ai dati QM;
il codice Pickyrecover , che recupera le energie e le geometrie intermolecolari dal database QM e una serie di altre utilità per assistere l’utente durante la parametrizzazione.
Gli FF intermolecolari, ottenuti con la procedura Picky, possono essere ulteriormente accoppiati con una parte intramolecolare per ottenere una descrizione più realistica del sistema. In tal caso, è possibile allestire un FF completamente derivato da QM adottando il protocollo Joyce per la parte intramolecolare.
Tutti i codici sono stati scritti in Fortran da Giacomo Prampolini, Ivo Cacelli, Antonella Cimoli e Paolo Roberto Livotto.
Tutto il software Picky è gratuito e può essere ridistribuito e/o modificato secondo i termini della GNU General Public License come pubblicata dalla Free Software Foundation (versione 3).
Download
Si prega di riempire il seguente form per ricevere il link per il download del software.
Manuale utente
Test suite e file di esempio
Applicazioni selezionate e parametri QMD-FF
Cristalli liquidi – predizione di proprietà del materiale tramite QMD-FF accurati
Topologie & files per Gromacs per cristalli liquidi
da Silveira, L.G., Livotto, P.R., Padula D., Vilhena, J.G., Prampolini, G. “Accurate Quantum-Mechanically Derived Force-Fields through a Fragment-Based Approach: Balancing Specificity and Transferability in the Prediction of Self-Assembly in Soft Matter”, J. Chem. Theor. and Comput., 18, 6905 (2022).
https://pubs.acs.org/doi/10.1021/acs.jctc.2c00747
Prampolini, G., da Silveira, L.G., Vilhena, J.G., Livotto, P.R “Predicting Spontaneous Orientational Self-Assembly: In Silico Design of Materials with Quantum Mechanically Derived Force Fields”, J. Phys. Chem. Lett., 13, 243 (2021).
https://pubs.acs.org/doi/10.1021/acs.jpclett.1c03517
Vilhena, J.G., da Silveira, L.G., Livotto, P.R., Cacelli, I., Prampolini, G. “Automated Parameterization of Quantum Mechanically Derived Force Fields for Soft Materials and Complex Fluids: Development and Validation”, J. Chem. Theor. and Comput., 17, 4449 (2021).
https://pubs.acs.org/doi/10.1021/acs.jctc.1c00213
Versioni precedenti (disponibili su richiesta):
Picky version 2.3 (route I and route II implementation, picky assistant automation, 2015, [2])
Picky version 2.5 (virtual sites implementation, placevirt utility, 2019, [3)]
Riferimenti
Sviluppo del metodo:
[1] I. Cacelli, A. Cimoli., P.R. Livotto, and G. Prampolini, G, “An automated approach for the parameterization of accurate intermolecular force-fields: Pyridine as a case study” J. Comp. Chem. , 33, 1055–1067 (2012)
[2] G. Prampolini, P.R. Livotto, and I. Cacelli, “Accuracy of Quantum Mechanically Derived Force-Fields Parameterized from Dispersion-Corrected DFT Data: The Benzene Dimer as a Prototype for Aromatic Interactions” J. Chem. Theory Comput., 11, 5182–5196 (2015)
[3] M. Campetella,N. De Mitri, and G. Prampolini “Automated parameterization of quantum-mechanically derived force-fields including explicit sigma holes: A pathway to energetic and structural features of halogen bonds in gas and condensed phase.” J. Chem. Phys., 153, 044106 (2020)
Applicazioni selezionate:
[4] G. Prampolini, M. Campetella, N. De Mitri, P.R. Livotto and I. Cacelli “Systematic and Automated Development of Quantum Mechanically Derived Force Fields: The Challenging Case of Halogenated Hydrocarbons.” J. Chem. Theor. Comput., 12, 5525–5540 (2016)
[5] L. Greff da Silveira, M. Jacobs, G. Prampolini, P.R: Livotto, and I. Cacelli “Development and Validation of Quantum Mechanically Derived Force-Fields: Thermodynamic, Structural, and Vibrational Properties of Aromatic Heterocycles” J. Chem. Theor. Comput., 14, 4884–4900 (2018)