
Joyce
An integrated and user-friendly tool for the parameterization of intra-molecular force fields from quantum mechanical data
The energy and its first and second geometrical derivatives obtained by Quantum Mechanical (QM) calculations for a number of conformations of a single molecule in its ground- or excited states are used to parameterize intra-molecular force fields, suitable for classical computer simulations.
A classical force-field (FF) is built up by essentially three ingredients, namely:
- A set of selected generalized (or redundant) internal coordinates (RIC), as bond lengths, angles, dihedrals or non-bonded distances, which completely define the molecular geometry;
- A set of model potential functions associated with each RIC;
- A set of parameters (force constants and RIC’s equilibrium values) which complete the definition of the model functions, settling molecular chemical specificity onto the FF functional.
As the first two points are regarded, Joyce, a FORTRAN77 program, reads a trial FF file in which both all the selected RIC and the associated model functions, which define the intra-molecular potential, are specified. This file can be automatically generated by the Joyce program, created by the user or built by standard MD software.
Currently, the supported file format for the input FF file is the .top GROMACS topology file.
The equilibrium values of the selected internal coordinates are read by the Joyce program from a formatted .fcc state file compatible with the FCClasse3.0 code or a .fchk checkpoint file produced by the GAUSSIAN16 package. Finally, the force constants are computed by the Joyce [1-3] procedure from the first and second derivatives read from aforementioned file containing the QM database.
Further information, tutorials and related tools are available at the Joyce dedicated gitlab website.
The Joyce program was written by Giacomo Prampolini, Javier Cerezo, Samuele Giannini, Nicola De Mitri and Ivo Cacelli.
This program is free software: you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation (version3).
For further information please contact Giacomo Prampolini, Javier Cerezo or Samuele Giannini.
Download
Please, fill the following form to receive the download link.
Installation and user guide
Free examples file
List of the references that should be cited in any article whose results are obtained, also partially, by using JOYCE:
Methods development:
[1] I. Cacelli and G. Prampolini, “Parametrization and Validation of of Intramolecular Force Fields Derived from DFT Calculations” J. Chem. Theory Comput., 3, 1803 (2007).
[2] V. Barone, I. Cacelli, N. De Mitri, D. Licari, S. Monti and G. Prampolini “JOYCE and ULYSSES: integrated and user-friendly tools for the parameterization of intra-molecular force fields from quantum mechanical data” Phys. Chem. Chem. Phys., 15, 3736 (2013).
[3] Cerezo, J., Prampolini, G. and Cacelli, I. Developing accurate intramolecular force fields for conjugated systems through explicit coupling terms. Theoretical Chemistry Accounts, 137, 80 (2018).
Selected application performed by the authors.
Organic dyes in solution – non phenomenological prediction of absorption spectral shapes
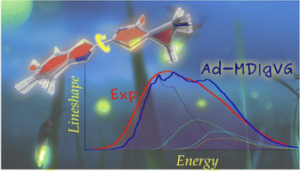
Cerezo, J., Garcìa-Iriepa, C., Santoro, F., Navizet, I., Prampolini, G. “Unraveling the contributions to the spectral shape of flexible dyes in solution: insights on the absorption spectrum of an oxyluciferin analogue.”, Phys. Chem. Chem. Phys., 23, 5007 (2023).
QMD-FF topologies & Gromacs related files
https://doi.org/10.1039/D2CP05701H
Intramolecular H-bond in solution : solvents effects on catechol binding affinity
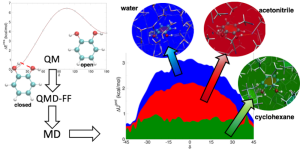
Ferretti, A., Campetella, M., Prampolini, G. “Solvent effects on catechol’s binding affinity: investigating the role of the intra-molecular hydrogen bond through a computational multi-level approach.”, Phys. Chem. Chem. Phys., 25, 2523 (2023).
QMD-FF topologies & Gromacs related files
https://doi.org/10.1039/D2CP04500A
Poly-pyrene – single molecule mechanical lifting
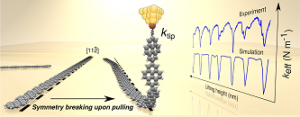
Pawlak, R., et al. “Sequential Bending and Twisting around C–C Single Bonds by Mechanical Lifting of a Pre-Adsorbed Polymer.” Nano Lett., 20, 652 (2020).
https://doi.org/10.1021/acs.nanolett.9b04418
Z907 – heteroleptic transition compound for DSSC: dynamical and environmental effects

Prampolini, G., Ingrosso, F., Cerezo, J., Iagatti, A., Foggi, P and Pastore, M. “Short- and Long-Range Solvation Effects on the Transient UV–Vis Absorption Spectra of a Ru(II)–Polypyridine Complex Disentangled by Nonequilibrium Molecular Dynamics.” J. Phys. Chem. Lett., 10, 2885 (2019).
https://doi.org/10.1021/acs.jpclett.9b00944
Prampolini, G., Ingrosso, F., Segalina, A., Caramori, S., Foggi, P. and Pastore, M. “Dynamical and Environmental Effects on the Optical Properties of an Heteroleptic Ru(II)–Polypyridine Complex: A Multilevel Approach Combining Accurate Ground and Excited State QM-Derived Force Fields, MD and TD-DFT.” J. Chem. Theor. and Comput., 15, 529 (2019).
https://doi.org/10.1021/acs.jctc.8b01031
Anthocyanidines – absorption properties and solvatochromism
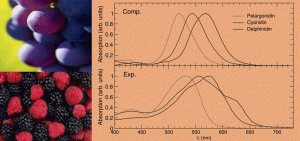
Cacelli, I., Ferretti, A., and Prampolini, G.. “Predicting light absorption properties of anthocyanidins in solution: a multi-level computational approach.” Theor. Chem. Acc., 135, 156 (2016).
https://doi.org/10.1007/s00214-016-1911-z
Fe-cyanides ions – 2D-IR spectroscopy for ions in solution

G. Prampolini, P. Yu, S. Pizzanelli, I. Cacelli, F. Yang, J. Zhao, and J. Wang “Structure and Dynamics of Ferrocyanide and Ferricyanide Anions in Water and Heavy Water: An Insight by MD Simulations and 2D IR Spectroscopy” J. Phys. Chem. B, 118 14899 (2014).
http://dx.doi.org/10.1021/jp511391b
Nfo-TEMPO – organic dye, solvated and grafted onto polymer matrices
N. De Mitri, S. Monti, G. Prampolini and V. Barone “Absorption and Emission Spectra of a Flexible Dye in Solution: a Computational Time-Dependent Approach” Chem. Theory and Comput., 9, 4507 (2013).
https://pubs.acs.org/doi/10.1021/ct4005799
G. Prampolini, S. Monti, N. De Mitri and V. Barone “Evidences of long lived cages in functionalized polymers: Effects on chromophore dynamic and spectroscopic properties” Chem. Phys. Lett., 601, 134 (2014).
http://www.sciencedirect.com/science/article/pii/S0009261414002632
N. De Mitri, G. Prampolini, S. Monti and V. Barone “Structural, dynamic and photophysical properties of a fluorescent dye incorporated in an amorphous hydrophobic polymer bundle” Phys.Chem.Chem.Phys., 16, 16573 (2014).
http://pubs.rsc.org/en/content/articlehtml/2014/cp/c4cp01828a
Tritc – organic dye, solvated and embedded in silica nanoparticles
V. Barone, J. Bloino, S. Monti, A. Pedone and G. Prampolini “Theoretical Multilevel Approach for studying the Photophysical Properties of Organic Dyes in solution” Phys. Chem. Chem. Phys., 12, 10550 (2010).
http://xlink.rsc.org/?DOI=c002722g
V. Barone, J. Bloino, S. Monti, A. Pedone and G. Prampolini “Fluorescence Spectra of Organic Dyes in Solution: A Time Dependent Multilevel Approach” Phys. Chem. Chem. Phys., 13, 2160 (2011).
http://xlink.rsc.org/?DOI=C0CP01320J
A. Pedone, G. Prampolini, S. Monti, and V. Barone “Realistic modeling of fluorescent dye-doped silica nanoparticles: A step toward the understanding of their enhanced photophysical properties” Chem. Mater., 23, 5016 (2011).
http://pubs.acs.org/doi/abs/10.1021/cm202436b