
FCclasses
Last available version is FCclasses 3.
FCclasses3 is a code written by Fabrizio Santoro (ICCOM-CNR) and Javier Cerezo (UAM) in Fortran 90 with some parts in Fortran 77. It computes vibronic spectra and nonradiative rates based on the harmonic approximation. It has been released in July 2022. Minor changes have been made on November 23 2022 (FCclasses 3.0.1) , on October 11 2023 (FCclasses 3.0.2) and on December 1 2023 (FCclasses 3.0.3)
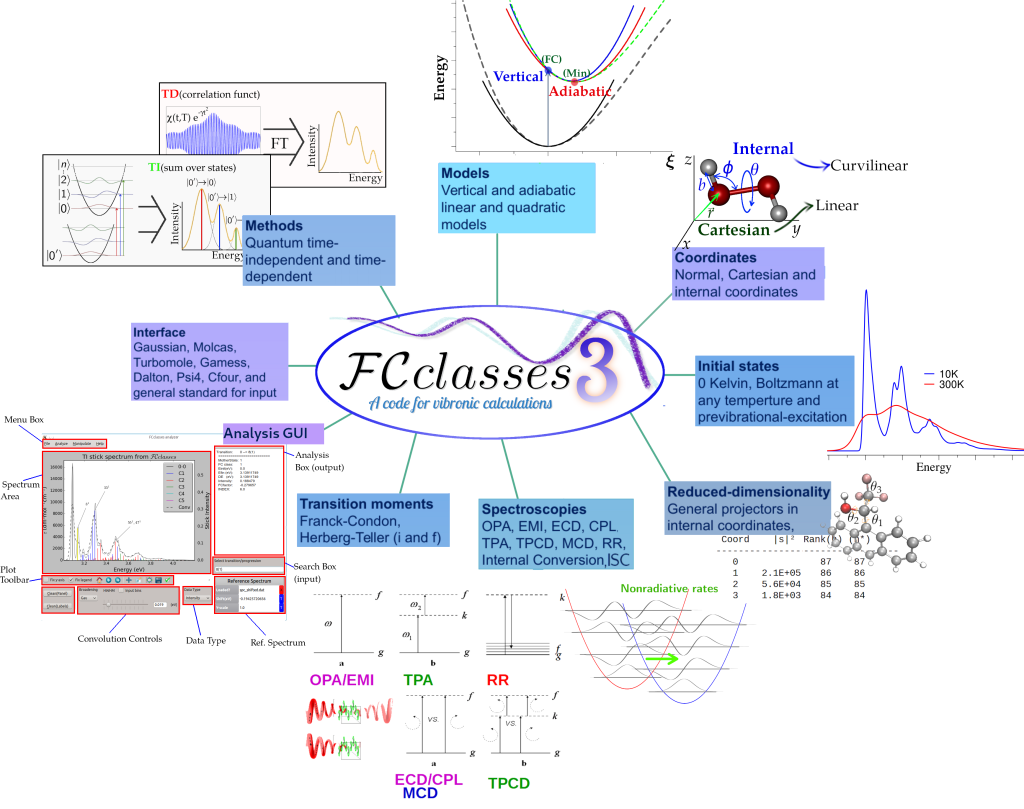
FCclasses 3.0 considerably extends the capabilities of the previous release FCclasses 2.1 with the possibility to compute a larger number of spectroscopies (one photon absorption, emission, electronic circular dichroism, circularly polarized luminescence, magnetic circular dichroism, two photon absorption, two photon circular dichroism and vibrational resonance Raman) and non radiative decays within Fermi golden rule framework (internal conversions triggered by nonadiabatic couplings, intersystem crossings and any process driven by a coupling that is a linear function of the nuclear coordinates). Concerning the model potential energy surfaces (PES), FCclasses 3.0 implements the full family of Vertical and Adiabatic harmonic models (AH, AS, ASF, VH, VG, VGF) and can use different sets of choice of coordinates (Cartesian and curvilinear internal coordinates, but also directly normal coordinates). The usage of curvilinear internal coordinates allows to define vibronic models expanded also in non-stationary points of the PES (generalized VH, gVH) and the availability of iterative projectors in internal coordinates allows to rigorously define reduced-dimensionality models. This feature is particularly useful to deal with flexible systems. All properties are implemented in both the time-independent (TI) sum-over-states approach through the pre-screening techniques elaborated in the previous version of the code, and a new time-dependent formulation, through the implementation of analytical time-correlation functions. These new features allows an efficient calculation at 0K and at any temperature. Specific initial states simulating a vibrational pre-excitation can also be defined.
Ease of use is now at the core of the development, with simple and flexible input structure, tools to facilitate the preparation of the data (from the output of several quantum chemistry codes) and graphical interfaces to analyze the results.
FCclasses 3.0 is the result of the work conducted in the group of Fabrizio Santoro at ICCOM during the last decade, and most of the features are already described in several papers (see publication list). The tools, also included in the package, constitute an independent subproject lead by Javier Cerezo, which can be accessed (and contributed) directly through its GitHub page
Changes in FCclasses 3.0.4 with respect to FCclasses 3.0.4 (july 2024)
- Fixed a bug introduced in the computation of nonradiative decay rates in combination with the time-dependent method
- Enabled the option to remove also normal coordinates of the final state RMCOOR=S2MODES
- Change fatal error to a simple warning for cases with non-negligible energy gradient in combination with AH model
- Improved some tools (check dedicated repository for details)
Changes in FCclasses 3.0.3 with respect to FCclasses 3.0.2 (december 2023)
- Fixed a bug introduced to IC rate calculations with time-dependent in the last release (causing some calculations to stop due to an error)
- Added a check on the generated spectra with respect reference ones (it works only if python with numpy and scipy is available)
- Few time-independent (TI) routines have been revised, solving some issues that might (rarely) lead to convergence problems or even segfaults (computations ended with a good convergence were surely ok)
- Added IR spectra as property (tools have been updated to support generation of derivative dipole files with gen_fcc_dipfiles)
Changes in FCclasses 3.0.2 with respect to FCclasses 3.0.1 (october 2023) - Fixed a bug in the data provided in file Bin_Spectrum.dat for properties different from OPA. Such file is useful to post-processing the spectrum computed with the time-independent approach and convolute it with a different linewidth - The reading of the file fcc is now subjected to less restrictions (like the number of spaces between different sections) - Added the possibility to compute the spectrum in semi-classical or classical Franck-Condon approximation by a proper sampling of the Wigner or Boltzmann distribution - Implemented the computation of the internal conversion rates with the time-dependent approach also at 0 Kelvin
Changes in FCclasses 3.0.1 with respect to FCclasses 3.0 (november 2022) - Fixed bug compiling with gfortran in Ubuntu 20.04 - Fixed the normalization of the TD lineshapes computed with internal coordinates and the AH model. Previous result is multiplied by |det(J)| where J is the Duschinsky matrix - RM_COORD option is now case insensitive - Added a check to stop an IC calculation with TD approach at 0K. In fact this option is not yet supported - Cartesian coordinates of the extrapolated minimum with the vertical model are now printed to the log file - Added rm_coord examples to the folder "test" - Removed checks on the D matrix. The information was not always meaningful. The analysis of the Duschinsky matrix is more useful. - Small modifications to the manual and tests -A tutorial is now available
For further information please contact Fabrizio Santoro or Javier Cerezo
Download (version 3.0.2, 11 october 2023)
Please, fill the following form to receive the download link.
User Manual and Tutorials (23 November 2022)
download the tutorial; download the data
Previous Versions of the code
If you are interested in obtaining the previous version of the code FCclasses 2.1 please contact directly Fabrizio Santoro. April M. Van Winkle and John W. Silzel at Biola University, modified the FCclasses 2.1 code to run it in parallel. For further information on this implementation please contact directly John W. Silzel.
References
[1] Javier Cerezo and Fabrizio Santoro, FCclasses3 , Vibrationally-resolved spectra simulated at the edge of the harmonic approximation, J. Comput. Chem. (2022) doi:10.1002/jcc.27027
[2] J. Cerezo, A. Aranda, F. J. Avila Ferrer, G. Prampolini, F. Santoro, Adiabatic-Molecular Dynamics Generalized Vertical Hessian Approach: A Mixed Quantum Classical Method to Compute Electronic Spectra of Flexible Molecules in the Condensed Phase, J. Chem. Theor. Comp. 16, 1215-1231, 2020
[3] A. Humeniuk, M. Bužčančić, J. Hoche, J. Cerezo, R. Mitrić, F. Santoro, V. Bonačić-Kouteckŷ, Predicting Fluorescence Quantum Yields for Molecules in Solution: A Critical Assessment of the Harmonic Approximation and the Choice of the Lineshape Function. J. Chem. Phys. 152, 054107, 2020
[4] J. von Cosel, J. Cerezo, D. Kern-Michler, C. Neumann, L. J. G. W. van Wilderen, J. Bredenbeck, F. Santoro, and I. Burghardt, Vibrationally resolved electronic spectra including vibrational pre-excitation: Theory and application to VIPER spectroscopy, J. Chem. Phys. 147, 164116, 2017
[5] J. Cerezo, F. Santoro Revisiting vertical models to simulate the line shape of electronic spectra adopting Cartesian and internal coordinates, J. Chem. Theor. Comp. 12, 4970-4985, 2016
[6] J. Cerezo, J. Zuniga, A. Requena, F. Avila Ferrer, F. Santoro, Harmonic models in Cartesian and internal coordinates to simulate the absorption spectra of carotenoids at finite temperatures, J. Chem. Theor. Comp. 9, 4947-4958, 2013.
[7] J. Cerezo, G. Mazzeo, G. Longhi, S. Abbate, F. Santoro, Quantum/Classical Calculation of Vibronic Spectra along a Reaction Path: The case of the ECD of Easily Interconvertible Conformers with Opposite Chiral Responses, J. Phys. Chem. Lett. 7, 4891-4897, 2016
[8] Y. Liu, J. Cerezo, G. Mazzeo, N. Lin, X. Zhao, G. Longhi, S. Abbate,F. Santoro, Vibronic coupling explains the different shape of electronic circular dichroism and of circularly polarized luminescence spectra of hexahelicene, J. Chem. Theor. Comp. 12, 2799-2819, 2016
[9] F. Avila, J. Cerezo, J. Soto, R. Improta, F. Santoro, First-principle computation of absorption and fluorescence spectra in solution accounting for vibronic structure, temperature effects and solvent inhomogenous broadening, Comp. Theor. Chem 1040-1041, 328-337, 2014
[10] N. Lin, H. Solheim, X. Zhao, F. Santoro, K. Ruud, First Principles Studies on the Vibrationally Resolved Magnetic Circular Dichroism Spectra of Biphenylene, J. Chem. Theor. Comp. 9, 1557-1567, 2013
[11] F. Avila, F. Santoro, Comparison of Vertical and Adiabatic Harmonic Approaches for the Calculation of the Vibrational Structure of Electronic Spectra, Phys Chem Chem Phys 14, 13549-13563, 2012
[12] F. Santoro, C. Cappelli, V. Barone, Efficient time-independent method for the calculation of resonance Raman spectra in sizeable molecules including Duschinsky and Herzberg-Teller effects, J. Comp. Theory and Comp 7, 1824-1839, 2011.
[13] F. Santoro, V. Barone, Computational approach to the study of the lineshape of absorption and electronic circular dichroism spectra, Int. J. Quantum. Chem 110, 624-636, 2010
[14] N. Lin, F. Santoro, A. Rizzo, Y. Luo, X. Zhao, , V. Barone, Theory for vibrationally resolved two-photon circular dichroism spectra. Application to (R)-(+)-3-methylcyclopentanone, J Phys. Chem A. 113, 4198-4207, 2009
[15] F. Santoro, R. Improta, A. Lami, J. Bloino, V.Barone, Effective method for the computation of optical spectra of large molecules at finite temperature including the Duschinsky and Herzberg-Teller effect. The Qx band of porphyrin as a case study, J. Chem. Phys. 128, 224311, 2008
[16] F. Santoro, R. Improta, A. Lami, V. Barone, An effective method to compute vibrationally resolved optical spectra of large molecules at finite temperature in the gas-phase and in solution, J. Chem Phys, 126, 184102, 2007
[17] F. Santoro, R. Improta, A. Lami, J. Bloino, V. Barone, Effective method to compute Franck-Condon integrals for optical spectra of large molecules in solution, J. Chem. Phys. 126, 084509, 2007.