
FCclasses
Versione distribuita FCclasses 3
FCclasses3 è un codice scritto da Fabrizio Santoro (ICCOM-CNR) e Javier Cerezo (UAM) in Fortran 90 con alcune parti in Fortran 77. Il codice calcola spettri vibronici e velocità di decadimento non radiativo a partire da modelli armonici . FCclasses 3.0 è stato distribuito a luglio 2022. Sono state introdotte delle modifiche minori e risolti dei piccoli problemi in data 23 novembre 2022 (FCclasses 3.0.1), in data 11 ottobre 2023 (FCclasses 3.0.2) e in data 1 dicembre 2023 (FCclasses 3.0.3).
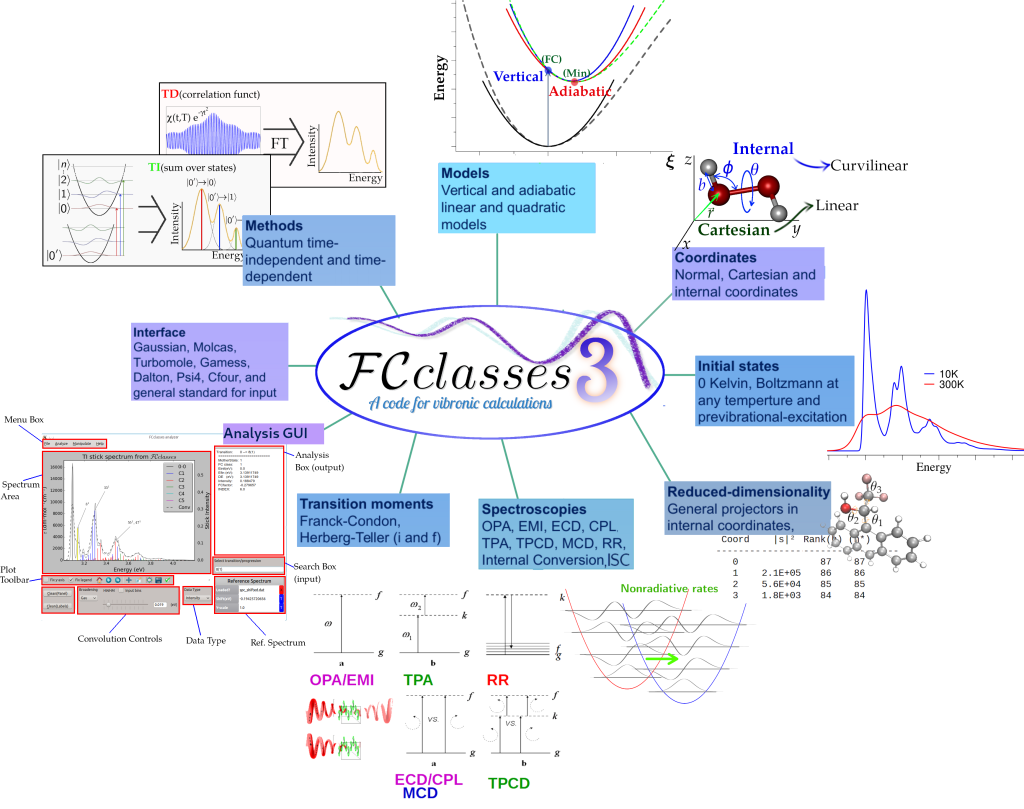
FCclasses 3.0 è una versione largamente riscritta del codice che estende significativamente le potenzialità della versione precedente FCclasses 2.1. FCclasses 3.0 fornisce ora la possibilità di calcolare un maggiore numero di spettroscopie (assorbimento ad un fotone, emissione, dicroismo circolare elettronico, luminescenza polarizzata circolarmente, dicroismo circolare magnetico, assorbimento a due fotoni, dicroismo circolare a due fotoni e Raman risonante vibrazionale) e velocità di decadimenti non radiativi descritti nell’ambito della regola d’ora di Fermi (conversione interne innescate da accoppiamenti non adiabatici, intersystem crossing e qualunque processo dovuto ad un accoppiamento che è una funzione lineare delle coordinate nucleari). Per quanto riguarda le surperfici di potenziale modello (PES), FCclasses 3.0 implementa la famiglia completa di modelli armonici Adiabatici (AH, AS, ASF) e Verticali (VH, VG e VGF) che consentono l’analisi dettagliata degli effetti di spostamenti, cambi di frequenza e rotazioni di Duschinsky, e può usare diversi insiemi di coordinate (coordinate Cartesiane, coordinate interne curvilinee, ma anche direttamente coordinate normali). L’uso di coordinate interne curvilinee consente la definizione di modelli verticali con espansioni di Taylor anche in punti non stazionali della PES (VH generalizzato, gVH) e l’implementazione di proiettori iterativi in coordinate interne da la possibilità di definire in maniera rigorosa modelli a dimensionalità ridotte. Ciò è particolarmente utile per descrivere sistemi flessibili. Tutte le proprietà possono essere calcolate sia con l’approccio indipendente dal tempo, somma-sugli-stati, grazie al metodo di pre-screening elaborato nelle versione precedenti del codice, sia con una nuova formulazione dipendente dal tempo, grazie alla implementazione di funzioni di correlazione analitiche. La nuova versione del codice dunque permette di svolgere efficientemente calcoli a qualsiasi temperatura. E’ anche possibile definire specifici stati iniziali, per simulare pre-eccitazioni vibrazionali.
La nuova versione del codice FCclasses 3.0 è stata progettata in modo da rendere il suo utilizzo il più semplice possibile, con una struttura dell’input semplice e flessibile e una serie di tools per la preparazione dei dati interfacciati con molti dei più popolari codici di chimica quantistica e interfacce grafiche per analizzare i risultati.
FCclasses 3.0 è il risultato del lavoro svolto nel gruppo di Fabrizio Santoro in ICCOM negli ultimi 10 anni e molte delle sue potenzialità sono già state descritte in pubblicazioni scientifiche (si guardi la lista dei riferimenti). Gli strumenti accessori, distribuiti con il pacchetto, fanno parte di un sotto-progetto indipendente, guidato da Javier Cerezo, che si può scaricare e a cui si può contribuire visitando la pagina GitHub
Per maggiori informazioni su FCclasses 3.0 si prega di contattare Fabrizio Santoro o Javier Cerezo
Modifiche in FCclasses 3.0.4 rispetto a FCclasses 3.0.3 (luglio 2024).
- Risolto un bug nel calcolo delle velocità di decadimento non radiativo con il metodo time-dependent
- Attivata la possibilità di rimuovere coordinate anche dello stato finale RMCOORD=S2MODES
- Modificato da fatal error ad un semplice warning il controllo per gradienti non negligibili in combinazione con il metodo AH
- Migliorati alcuni tools (visionare il repository dedicato per maggiori dettagli)
Modifiche in FCclasses 3.0.3 rispetto a FCclasses 3.0.2 (dicembre 2023).
- Corretto un errore introdotto in FCclasses 3.0.2 nel calcolo TD delle velocità di internal conversion (causa in alcuni casi di uno stop del calcolo)
- Inserito un controllo sugli spettri genrati rispetto a spettri di riferimento (funziona solo se è disponibile python con numpy e scipy)
- Risolto un problema in alcune routine del conti time-independent che poteva (raramente) causare segmentation faluts o problemi di convergenza durante il calcolo (calcoli terminati con buona convergenza sono sicuramente corretti)
- Apportate alcune modifiche all'output dei conti time-independent per renderli meno verbosi e aumentarne la leggibilità
- Implementato il calcolo degli spettri IR (i tools sono anche stati aggiornati per permettere la generazione dei file con le derivate del dipolo con gen_fcc_dipfiles)
Modifiche in FCclasses 3.0.2 rispetto a FCclasses 3.0.1 (ottobre 2023).
- Risolto un problema nei dati riportati nel file Bin_Spectrum.dat per proprietà diverse da OPA (il file è utile per ricalcolare spettri ottenuti con il metodo TI modificando l'allargamento fenomenologico) - La lettura dei file .fcc è ora più flessibile, cioè il formato di lettura è meno rigido per quanto riguarda ad esempio lo spazio tra le sezioni - Aggiunto il calcolo dello spettro in approssimazione classica di Franck Condon attraverso il sampling della distribuzione di Wigner (quantistica) o di Boltzmann (classica) - implementato il calcolo delle costanti di decadimento non radiativo con il metodo time-dependent anche alla temperatura di 0 Kelvin
Modifiche in FCclasses 3.0.1 rispetto a FCclasses 3.0 (novembre 2022).
- Risolto un problema nella compilazione con gfortran in Ubuntu 20.04 - Risolto un problema di normalizzazione delle lineshape spettrali calcolate in coordinate interne con il metodo AH. I risultati ottenuti con la versione precedente sono moltiplicati per il determinante della matrice di Duschinsky det(J)| - L'opzione RM_COORD ora è case insensitive - Aggiunto un controllo per impedire un calcolo IC con l'approccio time-dependent a 0K. Questa opzione non è ancora supportata - Per calcoli verticali, vengono ora stampate nel file di output le coordinate Cartesiane del minimo estrapolato dello stato finale - Sono stati aggiunti esempi di utilizzo della keyword rm_coord nel folder dei test - Non vengono più stampati dei checks sulla cosiddetta matrice D. L'informazione ottenuta non era infatti significativa in tutti i casi. E' più corretto basarsi sull'analisi della matrice di Duschinsky. - Sono state apportate piccole modifiche al manuale e ai test - Viene ora distribuito anche un tutorial all'utilizzo di FCclasses 3
Per maggiori informazioni contattare Fabrizio Santoro o Javier Cerezo
Download (versione dell’11 ottobre 2023)
Manuale (23 Novembre 2022)
Tutorial (23 novembre 2022): scarica le note; scarica i dati
Versione precedenti di FCclasses
Gli utenti interessati ad ottenere la versione precedente del codice FCclasses 2.1 sono pragati di contattare direttamente Fabrizio Santoro. April M. Van Winkle e John W. Silzel della Biola University, modificarono FCclasses 2.1 per eseguirlo in parallelo. Per ulteriori informazioni su questa implementazione, contattare direttamente John W. Silzel.
Riferimenti
[1] Javier Cerezo and Fabrizio Santoro, FCclasses3 , Vibrationally-resolved spectra simulated at the edge of the harmonic approximation, J. Comput. Chem. 44, 626-643, 2023
[2] J. Cerezo, A. Aranda, F. J. Avila Ferrer, G. Prampolini, F. Santoro, Adiabatic-Molecular Dynamics Generalized Vertical Hessian Approach: A Mixed Quantum Classical Method to Compute Electronic Spectra of Flexible Molecules in the Condensed Phase, J. Chem. Theor. Comp. 16, 1215-1231, 2020
[3] A. Humeniuk, M. Bužčančić, J. Hoche, J. Cerezo, R. Mitrić, F. Santoro, V. Bonačić-Kouteckŷ, Predicting Fluorescence Quantum Yields for Molecules in Solution: A Critical Assessment of the Harmonic Approximation and the Choice of the Lineshape Function. J. Chem. Phys. 152, 054107, 2020
[4] J. von Cosel, J. Cerezo, D. Kern-Michler, C. Neumann, L. J. G. W. van Wilderen, J. Bredenbeck, F. Santoro, and I. Burghardt, Vibrationally resolved electronic spectra including vibrational pre-excitation: Theory and application to VIPER spectroscopy, J. Chem. Phys. 147, 164116, 2017
[5] J. Cerezo, F. Santoro Revisiting vertical models to simulate the line shape of electronic spectra adopting Cartesian and internal coordinates, J. Chem. Theor. Comp. 12, 4970-4985, 2016
[6] J. Cerezo, J. Zuniga, A. Requena, F. Avila Ferrer, F. Santoro, Harmonic models in Cartesian and internal coordinates to simulate the absorption spectra of carotenoids at finite temperatures, J. Chem. Theor. Comp. 9, 4947-4958, 2013.
[7] J. Cerezo, G. Mazzeo, G. Longhi, S. Abbate, F. Santoro, Quantum/Classical Calculation of Vibronic Spectra along a Reaction Path: The case of the ECD of Easily Interconvertible Conformers with Opposite Chiral Responses, J. Phys. Chem. Lett. 7, 4891-4897, 2016
[8] Y. Liu, J. Cerezo, G. Mazzeo, N. Lin, X. Zhao, G. Longhi, S. Abbate,F. Santoro, Vibronic coupling explains the different shape of electronic circular dichroism and of circularly polarized luminescence spectra of hexahelicene, J. Chem. Theor. Comp. 12, 2799-2819, 2016
[9] F. Avila, J. Cerezo, J. Soto, R. Improta, F. Santoro, First-principle computation of absorption and fluorescence spectra in solution accounting for vibronic structure, temperature effects and solvent inhomogenous broadening, Comp. Theor. Chem 1040-1041, 328-337, 2014
[10] N. Lin, H. Solheim, X. Zhao, F. Santoro, K. Ruud, First Principles Studies on the Vibrationally Resolved Magnetic Circular Dichroism Spectra of Biphenylene, J. Chem. Theor. Comp. 9, 1557-1567, 2013
[11] F. Avila, F. Santoro, Comparison of Vertical and Adiabatic Harmonic Approaches for the Calculation of the Vibrational Structure of Electronic Spectra, Phys Chem Chem Phys 14, 13549-13563, 2012
[12] F. Santoro, C. Cappelli, V. Barone, Efficient time-independent method for the calculation of resonance Raman spectra in sizeable molecules including Duschinsky and Herzberg-Teller effects, J. Comp. Theory and Comp 7, 1824-1839, 2011.
[13] F. Santoro, V. Barone, Computational approach to the study of the lineshape of absorption and electronic circular dichroism spectra, Int. J. Quantum. Chem 110, 624-636, 2010
[14] N. Lin, F. Santoro, A. Rizzo, Y. Luo, X. Zhao, , V. Barone, Theory for vibrationally resolved two-photon circular dichroism spectra. Application to (R)-(+)-3-methylcyclopentanone, J Phys. Chem A. 113, 4198-4207, 2009
[15] F. Santoro, R. Improta, A. Lami, J. Bloino, V.Barone, Effective method for the computation of optical spectra of large molecules at finite temperature including the Duschinsky and Herzberg-Teller effect. The Qx band of porphyrin as a case study, J. Chem. Phys. 128, 224311, 2008
[16] F. Santoro, R. Improta, A. Lami, V. Barone, An effective method to compute vibrationally resolved optical spectra of large molecules at finite temperature in the gas-phase and in solution, J. Chem Phys, 126, 184102, 2007
[17] F. Santoro, R. Improta, A. Lami, J. Bloino, V. Barone, Effective method to compute Franck-Condon integrals for optical spectra of large molecules in solution, J. Chem. Phys. 126, 084509, 2007.