
Overdia
Overdia is a free open source code aimed at the definition of diabatic electronic states and the parametrization of Linear Vibronic Coupling (LVC) Hamiltonians from Time-Dependent Density Functional Theory calculations. It can be adopted to describe molecular potential energy surfaces close to conical intersections but it also parameterizes generalized LVC models for molecular dimers and aggregates, accounting for the existence of exciton and charge-transfer states and their competitions with internal conversion processes on the monomeric units. The code exploits a diabatization strategy based on the maximum overlap criterium. The generated LVC Hamiltonians can be employed to study the electronic nonadiabatic spectroscopy and the quantum dynamics of photoinduced processes. Overdia is expected to be useful to study the photophysical behaviour of molecules and molecular aggregates relevant in photochemistry and photobiology and/or for innovative soft materials with application, for instance, in organic photovoltaics and optoelectronics
Overdia is interfaced with the package Gaussian for electronic structure calculations and it automatically produces the necessary files to run quantum dynamical computations with the MCTDH code Quantics
Overdia was written in Fortran 90 by Fabrizio Santoro (ICCOM-CNR) and James Green (IBB-CNR)
The manual of Overdia 1.0, released on 4 March 2022 can be downloaded here. It includes a number of examples and short tutorials
Overdia is free software distributed under the GNU General Public License. It is distributed together with a number of small utilities to prepare the input files, analyse the diabatic potential energy surfaces and interface it with other programs. It is also possible to interface Overdia with other quantum chemistry packages. This has already be done to employ RASPT2 data obtained with MOLCAS. The interested users may contact the authors.
To download the release 1.0 of Overdia, released on 4 March 2022, you are kindly requested to fill the following form
Bibliography
The method for the parametrization of standard LVC Hamiltonians (where i.e. the references states for the definition of the diabatic states are taken identical to the adiabatic molecular states at a given molecular geometry) was reported here:
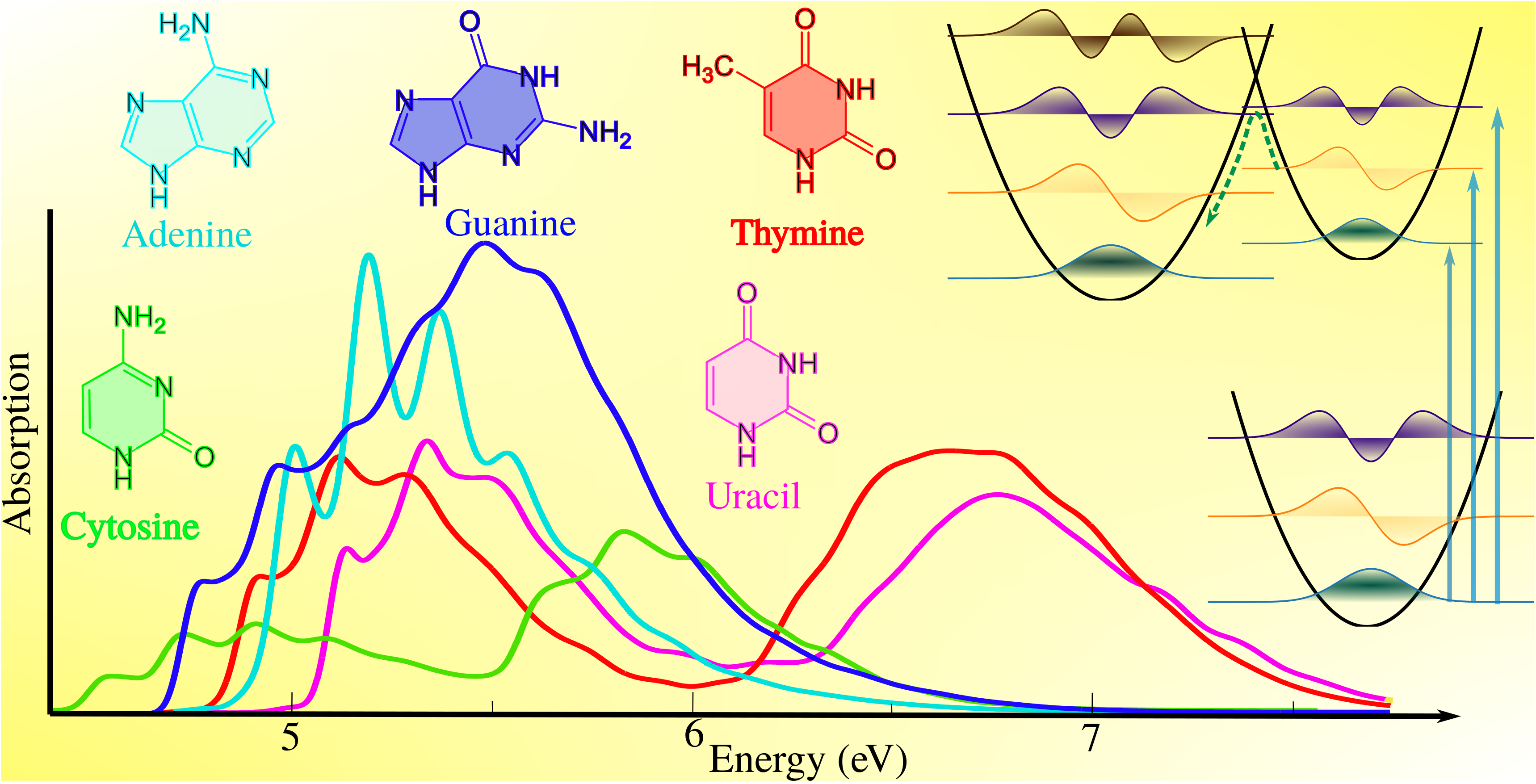 Martha Yaghoubi Jouybari, Yanli Liu, Roberto Improta, and Fabrizio Santoro, “Ultrafast dynamics of the two lowest bright excited states of cytosine and 1-methylcytosine: A quantum dynamical study”, Journal of Chemical Theory and Computation 16(9), pp. 5792–5808 (2020).
Martha Yaghoubi Jouybari, Yanli Liu, Roberto Improta, and Fabrizio Santoro, “Ultrafast dynamics of the two lowest bright excited states of cytosine and 1-methylcytosine: A quantum dynamical study”, Journal of Chemical Theory and Computation 16(9), pp. 5792–5808 (2020).
The single-point fragment diabatization method which also gives the possibility to the user to define custom diabatic states in terms of orbital transitions (even considering mixed molecular orbitals), adopted for instance, to define local excitations and charge trandfeer states and named FrDEX was presented in
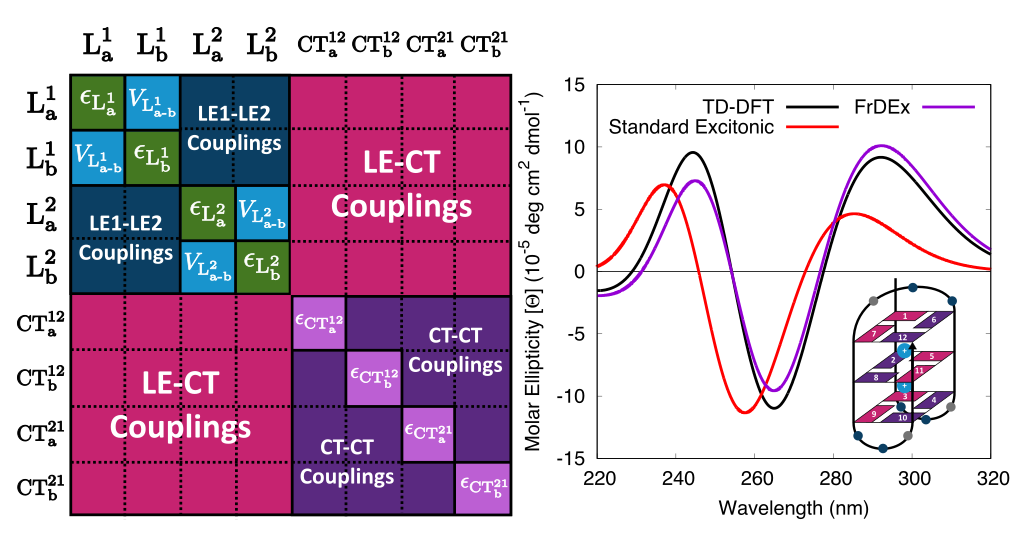 James A. Green, Haritha Asha, Fabrizio Santoro, and Roberto Improta, “Excitonic model for strongly coupled multichromophoric systems: The electronic circular dichroism spectra of guanine quadruplexes as test cases”, Journal of Chemical Theory and Computation 17(1), pp. 405–415 (2021)
James A. Green, Haritha Asha, Fabrizio Santoro, and Roberto Improta, “Excitonic model for strongly coupled multichromophoric systems: The electronic circular dichroism spectra of guanine quadruplexes as test cases”, Journal of Chemical Theory and Computation 17(1), pp. 405–415 (2021)
The method for building generalized LVC models based on the fragment diabatization was introduced and employed here:
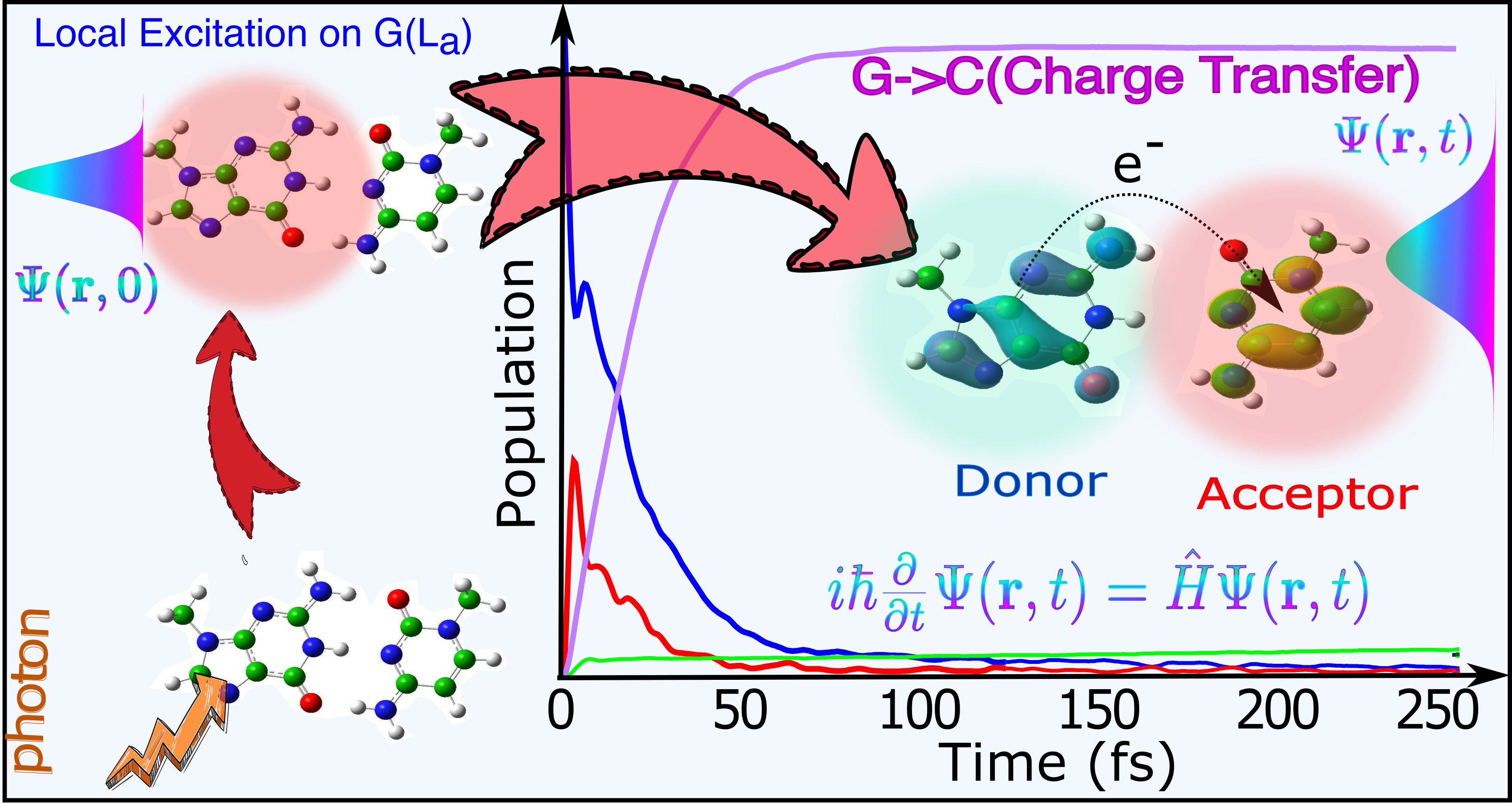 James A. Green, Martha Yaghoubi Jouybari, Haritha Asha, Fabrizio Santoro, and Roberto Improta, “Fragment diabatization linear vibronic coupling model for quantum dynamics of multichromophoric systems: Population of the charge-transfer state in the photoexcited guanine–cytosine pair”, Journal of
James A. Green, Martha Yaghoubi Jouybari, Haritha Asha, Fabrizio Santoro, and Roberto Improta, “Fragment diabatization linear vibronic coupling model for quantum dynamics of multichromophoric systems: Population of the charge-transfer state in the photoexcited guanine–cytosine pair”, Journal of
Chemical Theory and Computation 17(8), pp. 4660–4674 (2021)
Martha Yaghoubi Jouybari, James A. Green, Roberto Improta, and Fabrizio Santoro, “The ultrafast quantum dynamics of photoexcited adenine–thymine basepair investigated with a fragment-based diabatization and a linear vibronic coupling model”, The Journal of Physical Chemistry A 125(40), pp. 8912–8924 (2021)
A. Segalina, D. Aranda, J. A. Green, V. Cristino, S. Caramori, G. Prampolini, M. Pastore, F. Santoro “How the Interplay among Conformational Disorder, Solvation, Local, and Charge-Transfer Excitations Affects the Absorption Spectrum and Photoinduced Dynamics of Perylene Diimide Dimers: A Molecular Dynamics/Quantum Vibronic Approach”, Journal of Chemical Theory and Computation 2022, 18, 6, 3718-3736
A preliminary version of the code Overdia was adopted in 2018 in
Yanli Liu, Lara Martínez-Fernández, Javier Cerezo, Giacomo Prampolini, Roberto Improta, Fabrizio Santoro, Multistate coupled quantum dynamics of photoexcited cytosine in gas-phase: Nonadiabatic absorption spectrum and ultrafast internal conversions, Chem. Phys. 515, 452-463, 2018